In Campania sono stati diagnosticati 150 pazienti affetti dalla HHT, una malattia genetica rara che colpisce una persona ogni 5.000. Tra i sintomi più comuni si riscontra l’epistassi, ma la condizione può portare a conseguenze gravi come ictus e aneurismi.
Rara malattia HHT, in Campania diagnosticati 150 casi
L’HHT (Teleangectasia Emorragica Ereditaria), nota come “l’assassino silente”, è una malattia genetica rara che colpisce una persona ogni 5.000 nel mondo. Il sintomo più evidente è l’epistassi, ovvero la perdita di sangue dal naso, che può essere associata a molte altre condizioni. Tuttavia, l’HHT è molto più grave, poiché comporta una serie di malformazioni artero-venose in vari organi, come cervello, polmoni, fegato, reni e naso, a seconda della mutazione genetica coinvolta. La diagnosi è complessa.
In Campania, si stima che ci siano circa 150 pazienti diagnosticati, ma secondo l’associazione HHT Onlus Italia, il numero reale potrebbe superare il migliaio. “Molti non sono a conoscenza di essere affetti da questa malattia – affermano dall’associazione – e ci impegniamo a sensibilizzare l’opinione pubblica. Solo attraverso la prevenzione è possibile evitare le manifestazioni più gravi e le complicanze potenzialmente letali.”
HHT Onlus: “La Campania deve unirsi con i centri nazionali”
HHT Onlus è un’associazione che riunisce medici, pazienti, familiari, amici e sostenitori, tutti uniti dall’obiettivo di ridurre i tempi di diagnosi, combattere la solitudine attraverso l’ascolto, l’incontro e il supporto reciproco tra pazienti, costruire una rete nazionale di servizi e strutture operative e, infine, trovare una cura, che rappresenta la missione più importante. Domani, 28 febbraio, in occasione della giornata internazionale delle malattie rare, HHT Onlus Campania, che fa parte dell’associazione nazionale, organizzerà un evento significativo. “Nessuna malattia rara è più importante di un’altra – affermano dall’associazione – si tratta di un universo complesso e ancora poco conosciuto, in cui rientra anche l’HHT”.
Come si tratta? I centri di eccellenza principali si trovano per lo più nel nord Italia. Nel sud, il Policlinico di Bari si occupa di questa patologia. Tuttavia, le associazioni mirano a creare servizi territoriali efficienti che possano collaborare con i centri di eccellenza a livello nazionale e a stabilire procedure standardizzate per tutti i centri.
“Stiamo lavorando – afferma il coordinatore di HHT Onlus per la Regione Campania – per garantire che anche la Campania si integri nella rete dei centri di riferimento, permettendo così ai pazienti campani di effettuare screening e follow-up sul territorio. In collaborazione con il Professor Giuseppe Limongelli, docente associato di Cardiologia all’Università degli Studi della Campania e Direttore del Centro di Coordinamento Malattie Rare della Regione, stiamo formando un team di professionisti in grado di interagire efficacemente con i Centri di Eccellenza Italiani per l’HHT, come il Policlinico di Bari, il San Matteo di Pavia e l’Ospedale Maggiore di Crema. Puntiamo a raggiungere questo obiettivo entro la fine del 2025. Questi traguardi, sebbene ambiziosi, sono realizzabili solo attraverso un impegno collettivo e la sensibilità di tutti, anche di coloro che non sono direttamente coinvolti.”
Che cos’è l’HHT, una malattia genetica rara
L’HHT, che sta per Teleangectasia Emorragica Ereditaria e conosciuta anche come Sindrome di Rendu-Osler-Weber, è una malattia genetica rara che colpisce circa 1 persona su 5000 a livello globale, provocando malformazioni artero-venose. Il sintomo più comune è il sanguinamento dal naso, ma le anomalie che si possono riscontrare negli organi interni (come polmoni, cervello e fegato) possono portare a manifestazioni molto gravi, come ictus, ascessi cerebrali e aneurismi. Per questo motivo, la malattia è spesso definita “l’assassino silente”.
Nei pazienti affetti da HHT, si possono formare collegamenti diretti tra arterie e vene. Poiché manca la rete capillare, il flusso sanguigno non viene rallentato, e nel punto di giunzione tra arteria e vena, a causa della pressione elevata, i vasi tendono a diventare più sottili, aumentando così il rischio di sanguinamento. Inoltre, l’assenza della funzione di “filtro” dei capillari può portare a ulteriori gravi complicazioni.
La rarità della patologia, insieme a una serie di altre caratteristiche come il coinvolgimento di diversi organi e le sue manifestazioni insidiose, crea numerose difficoltà che complicano ulteriormente la diagnosi e il trattamento.
L’HHT è infatti una malattia ancora poco conosciuta e raramente diagnosticata: meno del 10% delle persone affette riceve una diagnosi, il che significa che solo 10 pazienti su 100 sono consapevoli della loro condizione. Il sanguinamento nasale è un sintomo frequentemente attribuito ad altre patologie più comuni e raramente viene considerato come un possibile segnale di questa malattia rara. Tuttavia, una diagnosi tardiva può esporre i pazienti a rischi significativi, con conseguenze potenzialmente gravi fin dall’infanzia.
A cosa serve lo screening precoce
L’importanza dello screening precoce diventa ancora più chiara considerando che attualmente non esistono cure farmacologiche o genetiche. Pertanto, adottare uno stile di vita sano, effettuare controlli regolari e intraprendere eventuali terapie sono essenziali per prevenire i danni causati dall’HHT. La malattia presenta diverse sfide impegnative per chi ne è colpito, che spesso si trova in difficoltà nel reperire informazioni, riferimenti e indicazioni, arrivando a sentirsi disorientato, sopraffatto e isolato.
“Il nostro impegno come associazione – dichiara il coordinatore di HHT Onlus per la Regione Campania – si sviluppa su vari fronti, ma il primo obiettivo che mi è stato assegnato è la diffusione della conoscenza della patologia, per identificare tutte le persone affette e garantire che si sottopongano allo screening preventivo.”
Siamo lieti di essere Charity Partner dal 2019 del rinomato Festival “Sui Sentieri degli Dei”, che si svolge lungo l’Alta Costiera amalfitana e ha visto la partecipazione di artisti di fama sia nazionale che internazionale. Grazie a questa iniziativa, volti noti del mondo della musica, del teatro, dell’arte e della letteratura hanno sostenuto la nostra associazione, contribuendo alla diffusione della conoscenza. Oltre a realizzare campagne informative, abbiamo anche trovato imprenditori disposti a sostenere la nostra causa, donando borse di studio per supportare una nuova generazione di professionisti e medici impegnati nella lotta contro l’HHT.
I sintomi e come si manifesta la rara malattia HHT
In ogni paziente affetto da H.H.T., la maggior parte dei vasi sanguigni risulta completamente normale, mentre solo una piccola parte presenta anomalie specifiche. In generale, i vasi sanguigni hanno la funzione di trasportare il sangue all’interno del corpo. Il cuore pompa il sangue con una certa pressione attraverso le arterie verso il resto dell’organismo, mentre le vene raccolgono il sangue dalla periferia e lo riportano, a bassa pressione, al cuore. Normalmente, arterie e vene non sono direttamente collegate; questa connessione avviene tramite piccoli vasi chiamati “capillari”. La H.H.T. causa un difetto nello sviluppo della rete vascolare, portando il sangue a passare direttamente da un’arteria al circuito venoso, senza il passaggio intermedio dei capillari. A causa della pressione esercitata dal sangue arterioso, la parete della vena si gonfia e diventa fragile, aumentando il rischio di rottura e conseguente emorragia.
In base alla tipologia dell’anomalia vascolare, essa può essere classificata come “teleangiectasia”, “fistola” o “malformazione artero-venosa” (MAV). Le teleangiectasie tendono a colpire principalmente le superfici interne ed esterne del corpo, interessando in particolare la pelle e le mucose, con un’incidenza elevata nella mucosa nasale. Al contrario, le fistole e le MAV si manifestano più frequentemente negli organi interni. In generale, le aree maggiormente interessate dalle lesioni vascolari associate alla H.H.T. includono il naso, la cavità orale, il volto, le mani, le mucose dello stomaco e dell’intestino, oltre al fegato, ai polmoni e al cervello. Tuttavia, non è chiaro il motivo di questa distribuzione preferenziale.
Le teleangiectasie e le malformazioni artero-venose (MAV) possono manifestarsi in modi diversi a seconda della zona del corpo interessata. La conseguenza più comune è il sanguinamento, che si verifica soprattutto a livello del naso, della pelle, dello stomaco e dell’intestino. Le MAV localizzate nel cervello e nel fegato, essendo raramente soggette a sanguinamento, risultano più difficili da individuare e richiedono quindi una competenza specifica da parte del medico che segue il paziente affetto da H.H.T. È importante sottolineare che è praticamente impossibile osservare, in un singolo paziente, un quadro clinico completo di tutte le lesioni e dei sintomi potenziali: una caratteristica distintiva della malattia è infatti la sua notevole variabilità clinica, anche tra i membri di una stessa famiglia. A volte, un genitore affetto può presentare epistassi molto gravi senza MAV negli organi interni, mentre il figlio può avere meno episodi di epistassi ma manifestare MAV in uno o più organi.
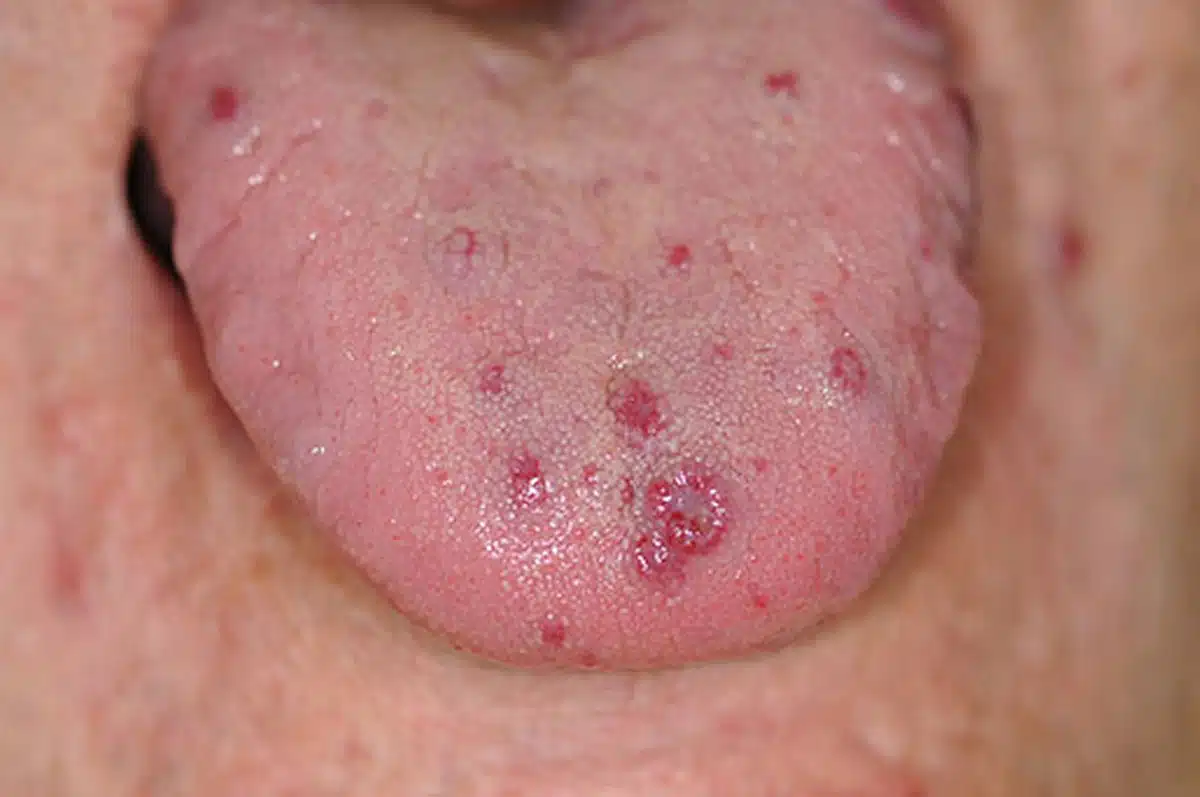
Non esiste un legame diretto tra il numero di teleangiectasie, la frequenza delle epistassi e il rischio di sviluppare malformazioni artero-venose (MAV) interne. In altre parole, chi presenta poche epistassi ha lo stesso rischio di sviluppare MAV polmonari, ad esempio, di chi soffre di frequenti sanguinamenti nasali. Le epistassi causate da teleangiectasie nasali sono il sintomo più comune della malattia, riscontrato nell’80-95% dei pazienti adulti, e tendono a manifestarsi mediamente a partire dai 12 anni, sebbene possano anche comparire in età infantile. La loro frequenza può variare da una o due volte all’anno fino a più volte al giorno, con una durata che va da pochi secondi a diverse ore. La quantità di sangue perso può variare da poche gocce a perdite così abbondanti da richiedere una trasfusione. Nella maggior parte dei casi, la gravità delle epistassi si colloca in una fascia intermedia tra questi due estremi.
Molti individui presentano anche teleangiectasie sulla pelle delle mani (soprattutto sui polpastrelli), sul volto e sulle mucose della cavità orale, che tendono a manifestarsi più tardi, generalmente tra i 30 e i 50 anni. Queste lesioni sono piccole, rilevate e molto fragili, con un colore che varia dal rosso al violaceo, di dimensioni simili a quelle di una capocchia di spillo o leggermente più grandi, formate da capillari dilatati e intrecciati. In alcuni casi, le teleangiectasie sono particolarmente evidenti, soprattutto in età adulta, mentre in altri rimangono meno numerose; tuttavia, con l’avanzare dell’età, tendono a diventare più frequenti. Circa il 40% dei pazienti può sviluppare una o più fistole polmonari, compromettendo una delle funzioni vitali normalmente svolte dalla rete capillare polmonare. Questo può portare a sintomi come pallore, affaticamento e vertigini, segni di una progressiva anemia.
Quando una fistola raggiunge un certo diametro, essa ostacola la capacità dei capillari polmonari di trattenere le impurità presenti nel sangue, come piccoli coaguli, colonie batteriche e microscopiche bolle d’aria. Senza questo filtro, tali impurità possono entrare nel sangue arterioso e, attraverso il cuore, dirigersi principalmente verso il cervello, causando così un ictus o un ascesso cerebrale, rispettivamente a causa di un coagulo o di un’infezione batterica. È meno comune, ma possibile, che si verifichi un sanguinamento da queste fistole, con un rischio leggermente maggiore per le donne in gravidanza. Queste complicanze sono sempre gravi e possono essere potenzialmente letali, o comunque portare a significative disabilità. Tuttavia, le fistole polmonari possono sempre essere trattate in modo radicale.
Nel 50-70% dei pazienti affetti, si osservano fistole all’interno del fegato, che possono causare scompensi cardio-circolatori ed epatici, soprattutto quando di dimensioni maggiori. Il flusso sanguigno anomalo che attraversa queste fistole provoca un sovraccarico di lavoro per il cuore e un aumento della pressione nel sistema venoso portale, responsabile della delicata “nutrizione” del fegato. In generale, il trattamento delle fistole epatiche non è necessario e viene riservato ai casi in cui esse causano le complicanze menzionate. Le malformazioni vascolari cerebrali (CAVM) sono state riscontrate nell’8-10% dei casi e, sebbene di solito siano asintomatiche, in una minoranza di situazioni possono portare a sanguinamenti letali o invalidanti; tuttavia, anche queste possono essere trattate in modo efficace nella maggior parte dei casi.
Le MAV a livello del midollo spinale sono meno comuni e generalmente trattabili. Possono essere sospettate in caso di forti dolori irradianti dalla colonna vertebrale o di disturbi della sensibilità in un braccio o in una gamba. Circa il 30% delle persone affette da morbo di Rendu-Osler presenta sanguinamenti nel tratto gastrointestinale, che possono variare da lievi a gravi, causati dalla formazione di teleangiectasie nella mucosa dello stomaco e dell’intestino, simili a quelle presenti sulla pelle. Le lesioni più a rischio di rottura si trovano nello stomaco e nella parte iniziale dell’intestino tenue, dove l’emorragia può manifestarsi con la presenza di sangue rosso nelle feci o con feci di aspetto particolare.
La cura
Sebbene non sia possibile prevenire le lesioni vascolari tipiche della malattia, nella maggior parte dei casi è possibile curarle. È importante intervenire solo quando queste causano disturbi significativi (ad esempio, epistassi frequenti) o quando comportano un elevato rischio di complicanze (come l’ictus cerebrale da fistola polmonare). Le opzioni terapeutiche variano a seconda della posizione e delle dimensioni delle lesioni. Per ridurre le epistassi, ad esempio, a volte sono sufficienti semplici accorgimenti quotidiani, come mantenere un buon livello di umidità nell’ambiente domestico e utilizzare pomate emollienti a livello nasale, che aiutano a diminuire la fragilità delle teleangiectasie. Quando queste misure non sono più efficaci, il trattamento inizialmente consigliato è la fotocoagulazione laser, in cui il raggio luminoso viene diretto non sulla lesione stessa, ma attorno ai suoi bordi, portando alla sua scomparsa.
I benefici ottenuti sono spesso temporanei, rendendo necessario ripetere il trattamento a intervalli regolari. È fondamentale rivolgersi sempre a specialisti esperti nell’uso del laser in ambito otolaringoiatrico. La dermoplastica del setto rappresenta un’ulteriore opzione terapeutica, da considerare solo dopo che il laser ha dimostrato di non essere efficace in più occasioni. Questa delicata procedura chirurgica prevede l’asportazione della sottile mucosa che riveste la cavità nasale, insieme alle teleangiectasie sanguinanti, sostituendola con una porzione di cute più resistente. Per ottenere risultati ottimali e duraturi, il paziente deve impegnarsi a mantenere la cavità nasale sempre umida e priva di secrezioni. In rari casi, può verificarsi una recidiva, ma in tal caso non è possibile ripetere l’intervento e sarà necessario adottare altre strategie di trattamento.
L’embolizzazione arteriosa è una procedura che può contribuire a ridurre le epistassi resistenti ad altre terapie, ma la sua efficacia è temporanea, poiché le arterie collaterali alla zona embolizzata possono portare a una recidiva nel tempo. Pertanto, questa tecnica è particolarmente indicata per affrontare situazioni di emergenza. Un trattamento ormonale somministrato per via intramuscolare può risultare efficace in alcune pazienti di sesso femminile che non rispondono né alle misure di umidificazione nasale né ai trattamenti di fotocoagulazione laser. Farmaci antiangiogenici, come il bevacizumab e la talidomide, hanno dimostrato di ridurre l’intensità delle epistasi e dell’anemia nei casi più gravi, ma la durata della loro efficacia e la loro sicurezza non sono ancora completamente chiarite.
La laserterapia dermatologica è efficace nel trattamento delle teleangectasie cutanee che presentano sanguinamenti significativi o che risultano particolarmente antiestetiche. In questi casi, è consigliabile consultare un esperto in laser dermatologici. Le emorragie gastrointestinali devono essere affrontate poiché possono causare anemia. Si inizia con integratori orali di ferro, ma nei casi più gravi si ricorre a emotrasfusioni e a trattamenti di cauterizzazione termica o laser tramite endoscopia. Anche la terapia ormonale e i farmaci antiangiogenici possono offrire benefici, sebbene con alcune riserve. Le fistole polmonari, invece, devono essere trattate prima che provochino disturbi neurologici o emorragie; pertanto, è importante cercarle anche nei pazienti asintomatici. Il trattamento iniziale prevede un’embolizzazione, che può essere eseguita in day hospital da un radiologo interventista.
Le cause
La malattia è provocata da diverse mutazioni in uno dei due geni finora identificati, situati rispettivamente sul cromosoma 9 e sul cromosoma 12. Il primo gene è responsabile della sintesi dell’endoglina, mentre il secondo codifica per una proteina nota come ALK-1. Ogni paziente, così come i membri della sua famiglia affetti da H.H.T., presenta la stessa mutazione in uno dei due geni. L’endoglina e l’ALK-1 sono proteine fondamentali per il corretto sviluppo dei vasi sanguigni; un’anomalia in uno di questi geni porta a un difetto nella produzione della proteina corrispondente, compromettendo così lo sviluppo dei vasi sanguigni.
La H.H.T. è una malattia genetica di tipo “autosomico dominante”. Questo implica che ogni persona affetta presenta, nel gene specifico, un allele normale e uno mutato. Durante ogni gravidanza, un genitore affetto da H.H.T. ha una probabilità del 50% di trasmettere al proprio bambino la mutazione e, di conseguenza, la malattia, senza alcuna distinzione di sesso. Se il bambino eredita la mutazione, non è possibile prevedere l’entità della gravità clinica della malattia. Al contrario, se il figlio riceve entrambi gli alleli normali, non solo non svilupperà la malattia, ma questa non sarà trasmessa alle generazioni future.
È quindi impossibile che la malattia salti generazioni, poiché per questo tipo di trasmissione ereditaria non esistono “portatori sani” in grado di trasmetterla senza esserne affetti. Se una persona con H.H.T. non ha genitori malati, ciò potrebbe essere dovuto alla particolare benignità del decorso della malattia nel genitore, tale da non destare alcun sospetto diagnostico. Quando si scopre un nuovo caso di H.H.T., è fondamentale, se possibile, esaminare attentamente entrambi i genitori per confermare ulteriormente la diagnosi e prevenire eventuali complicanze. È importante notare che, sebbene raro, un soggetto con H.H.T. può avere entrambi i genitori sani a causa di una mutazione casuale e inaspettata in uno dei geni coinvolti, avvenuta nelle prime fasi del concepimento (definita “neomutazione”). Tuttavia, nella maggior parte dei casi, la mutazione è già presente negli antenati da diverse generazioni.